TSENAT is a R package for quantifying and modeling isoform-usage complexity across RNA-seq samples using Tsallis entropy - a scale-dependent information-theoretic measure of transcript heterogeneity.
The Problem
Standard differential expression tools (DESeq2, edgeR) detect changes in total transcript abundance. However, genes often reorganize their isoform diversity without changing total abundance: they may shift from a balanced isoform distribution to dominance by a single isoform, or vice versa. This isoform switching and splicing-driven regulation is biologically important for cell state and function but invisible to abundance-focused methods.
The Solution
TSENAT captures isoform diversity independently of which specific isoforms are abundant. The method uses Tsallis entropy, whose entropic index parameter (q) enables to evaluate different data dimensions:
Low q (e.g., 0.5): Focuses on rare isoforms - detects if diversity is maintained or collapsed
Mid q (e.g., 1.0): Balanced view (Shannon entropy) - overall isoform complexity
High q (e.g., 2.0): Focuses on dominant isoforms - detects dominance shifts
By examining diversity across multiple entropic indices (q-values), you identify scale-dependent diversity changes - the hallmark of coordinate isoform switching.
The Mathematics Behind Tsallis Entropy
Tsallis entropy is a parametric family of diversity measures that generalizes Shannon entropy and enables tuning sensitivity to different scales of isoform organization.
Mathematical Definition: For a discrete probability vector representing isoform proportions within a gene, Tsallis entropy is:
This elegant formula unifies diverse diversity concepts at specific entropic indices (q-values):
- q = 0: Richness — Simple count of expressed isoforms; emphasizes rare variants most strongly.
- q = 1: Shannon entropy — Standard information-theoretic measure; balanced weighting across scales.
- q = 2: Gini-Simpson index — Probability that two randomly-drawn transcripts are different; robust to rare variants.
Divergence Analysis: Measuring Information-Theoretic Distance Between Conditions
While Tsallis entropy quantifies diversity within a single distribution, Tsallis divergence measures the information-theoretic distance between two distributions.
Mathematical Definition (Furuichi formula): For two probability distributions and representing isoform proportions in control and treatment conditions, Tsallis divergence is:
where and are the probability values at position .
Tsallis divergence enables the quantification of how fundamentally different the isoform complexity patterns are between experimental conditions.
Installation
Requirements: R >= 4.5.0
The easiest way to install TSENAT is through Bioconda:
To install the latest development version from GitHub:
remotes::install_github("gallardoalba/TSENAT")Quick Start
Load Example Data
Start by loading the built-in example dataset from TSENAT, which includes transcript-level read counts, TPM values, and effective lengths from Salmon quantification. Then load the sample metadata and annotation file that describe your experimental design.
suppressMessages({
library(TSENAT)
library(SummarizedExperiment)})
# Load example dataset (includes readcounts, tpm, and effective_length)
data(readcounts)
readcounts <- as.matrix(readcounts)
# Load sample metadata and annotation
metadata_df <- read.table(
system.file("extdata", "metadata.tsv", package = "TSENAT"),
header = TRUE,
sep = "\t")
gff3_file <- system.file(
"extdata", "annotation.gff3.gz", package = "TSENAT")Create configuration and build analysis
Create a configuration object specifying your experimental design parameters (sample/condition columns from metadata) before building the analysis object. This fail-fast pattern ensures invalid parameters are caught immediately before processing begins.
## Create configuration file
config <- TSENAT_config(
q = seq(0, 2, by = 0.05),
sample_col = "sample",
condition_col = "condition",
paired = TRUE,
subject_col = "paired_samples",
control = "normal",
nthreads = 2
)
## Build TSENATAnalysis object
analysis <- build_analysis(
readcounts = readcounts,
tx2gene = gff3_file,
metadata = metadata_df,
config = config,
tpm = tpm,
effective_length = effective_length)Orchestration Function
The TSENAT() function provides a complete, automated analysis pipeline in a single call. It takes your configured TSENATAnalysis object and executes all downstream analysis steps: entropy computation, statistical testing for entropic index (q-value) by condition interactions, and rich visualization. This is the recommended entry point for most users—it orchestrates the full workflow while respecting your configuration parameters (entropic indices, design, bootstrap settings, etc.) and handles output management seamlessly.
# Returns: Fully configured TSENATAnalysis object
result <- TSENAT(analysis)Accessing Results
After running the analysis, retrieve results using the unified results() interface:
# Unified interface: request results by type
diversity_results <- results(result, type = "diversity")
sait_results <- results(result, type = "sait")
jackknife_results <- results(result, type = "jackknife", q = 1)
# Filter and rank results flexibly
ranked_sait <- results(result, type = "sait", rankBy = "padj", n = 50)
filtered_diversity <- results(result, type = "diversity", q = 1.0)
# Retrieve plots
diversity_plot <- results(result, type = "diversity", plot=TRUE)
print(diversity_plot)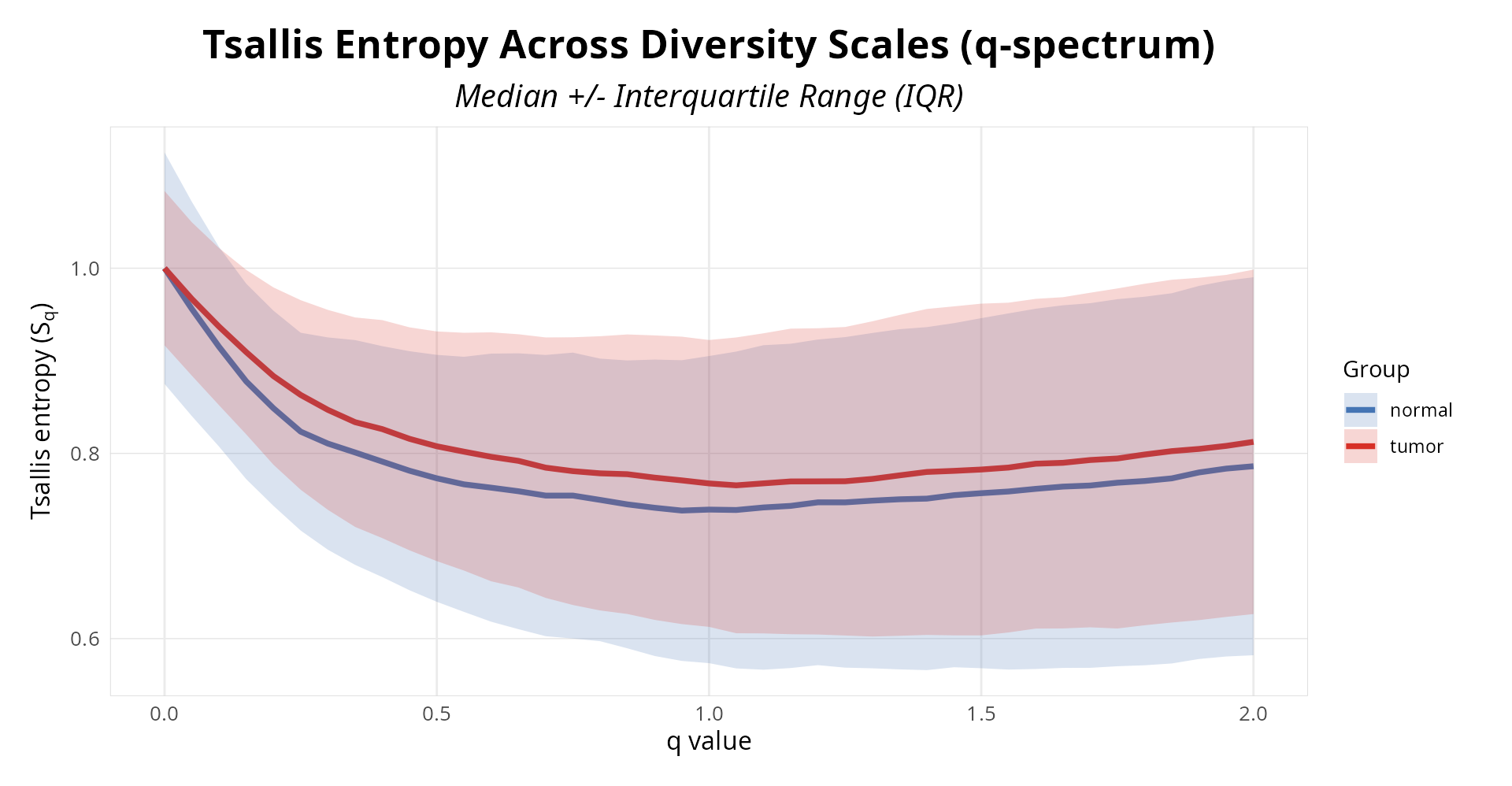
For a complete walkthrough of the analysis pipeline with real biological examples, see the main package vignette. This includes theory background, step-by-step explanations of each analysis function, and interpretation guidance for understanding your results.
Statistical Inference Methods
TSENAT provides a flexible statistical framework optimized for entropy-based diversity analysis. The recommended main workflow relies on Generalized Additive Models (GAM) via mgcv::gam() combined with ARIMA differencing.
The statistical methods available in TSENAT include:
- Scale-adaptive interaction tests (SAIT): Multiple modeling approaches optimized for repeated measures with AR(1) correlation structure. GAM, LMM, GEE and FPCA are all parametrized to handle the non-normality and heteroscedasticity characteristic of entropy data.
- Scheirer-Ray-Hare (SRH) rank tests: The test operates solely on ranks, making it robust to outliers and extreme values. However, it shows lower power for interactions and can inflate Type I errors under heterogeneous variance. These limitations are mitigated through Hochberg multiple testing correction.
- M-estimation: Robust location estimation for group comparison using iteratively re-weighted least squares, resistant to outliers.
- Jackknife isoform switching (JIS): Leave-one-out resampling to identify transcripts with condition-specific switching patterns and quantify their influence on entropy differences.
Related Packages
Below is how TSENAT complements other Bioconductor tools:
| Tool | Answers | TSENAT Difference |
|---|---|---|
| DESeq2, edgeR, limma | Which genes change in total abundance? | TSENAT detects isoform-usage complexity changes independent of total abundance |
| DRIMSeq | Which individual transcripts shift usage? | TSENAT measures overall isoform diversity, not individual transcript shifts |
| IsoformSwitchAnalyzeR | Which individual isoforms switch; what are the functional consequences? | TSENAT measures overall isoform diversity and diversity shifts rather than cataloging individual transcript switches or predicting functional consequences; complements switch identification with diversity patterns |
| SplicingFactory | What is the overall isoform diversity? | TSENAT extends with scale-dependent diversity (q-spectrum) vs fixed measures |
| Kallisto, Salmon | How many reads per transcript? | TSENAT uses their quantification as input; adds diversity analysis layer |
Native Salmon Integration
TSENAT is specifically engineered to work seamlessly with Salmon quantification output. Rather than requiring manual parsing or format conversion, TSENAT automatically discovers transcript-level quantification files across your Salmon output directory and integrates them directly into the analysis pipeline. This tight integration means you can move from Salmon quantification to entropy analysis without intermediate data manipulation—the raw quant.sf files are all you need. TSENAT discovers these files automatically, validates their compatibility with your experimental design, and handles length-correction and normalization as part of the diversity computation workflow.
To get started with Salmon-quantified data:
suppressMessages(library(TSENAT))
# Prepare configuration FIRST
config <- TSENAT_config(
q = seq(0, 2, by = 0.1),
condition_col = "condition",
sample_col = "sample",
subject_col = "paired_samples"
)
# Build analysis directly from Salmon output directory
analysis <- build_analysis(
salmon_dir = "path/to/salmon_output", # Auto-discovers all quant.sf files
tx2gene = "path/to/annotation.gff3.gz", # Transcript-to-gene mapping
metadata = metadata_df, # Sample metadata (see structure below)
config = config
)
# Run analysis pipeline
analysis <- TSENAT(analysis)Salmon Directory Structure
Your Salmon output must be organized with one folder per sample, each containing a quant.sf file with transcript-level quantification. Sample folder names must exactly match the row names in your metadata file (case-sensitive) to ensure correct sample attribution.
TSENAT expects Salmon output organized with one subdirectory per sample:
salmon_output/
├── Sample_1/
│ └── quant.sf
├── Sample_2/
│ └── quant.sf
├── Sample_3/
│ └── quant.sf
└── Sample_4/
└── quant.sfMetadata File Structure
Expected TSV file format:
sample condition paired_samples
SRR14800481 normal A
SRR14800480 normal B
SRR14800479 tumor A
SRR14800478 tumor BKey requirements:
-
samplecolumn: must match Salmon folder names exactly. -
conditioncolumn: experimental groups. -
paired_samplescolumn: required if using paired designs; identifier for matched samples.
Tests coverage
Testing is vital in research as it ensures the validity and reliability of results, which is essential for accurately interpreting findings. The report about the current testing coverage can be found here.
Package Architecture
Get a visual overview of TSENAT’s structure in package architecture. This helps you understand how the 600+ functions that integrate TSENAT work together.
License and Attribution
This project is licensed under the GNU General Public License v3.0 (GPL-3). See LICENSE for details.
TSENAT builds upon the SplicingFactory package, extending it with specialized focus on Tsallis entropy analysis.
“If I ever come back from the past, it’s to create a cyclone.”
– Juan José Lozano